
LLC et Richter – synthèse des recommandations EHA 2026
Leucémie lymphoïde chronique & transformation de Richter
Synthèse des recommandations EHA 2026 (Eichhorst & Ghia, HemaSphere 2026;10:e70403)
1. Introduction et méthodologie
Première édition rédigée sous l’égide de l’EHA (en accord avec l’ESMO), qui sera actualisée annuellement, avec un périmètre élargi à une prise en charge globale : du diagnostic et de la surveillance active jusqu’à la transformation de Richter, désormais nommée dans le titre. Intégration d’experts en diagnostic moléculaire et stratification pronostique, et participation de représentants de patients.
Double système de cotation : niveau de preuve (I = ≥1 grand essai randomisé de qualité ou méta-analyse homogène ; II = petits essais ou suspicion de biais ; III = cohortes prospectives ; IV = cohortes rétrospectives/cas-témoins ; V = sans contrôle, avis d’experts) et grade de recommandation (A = preuve forte + bénéfice substantiel, fortement recommandé ; B = preuve forte/modérée mais bénéfice limité ; C = preuve insuffisante, optionnel ; D = généralement non recommandé ; E = jamais recommandé). Première utilisation des scores MCBS:H : un score de 4 ou 5 indique un bénéfice substantiel en situation non curative.
2. Incidence et épidémiologie
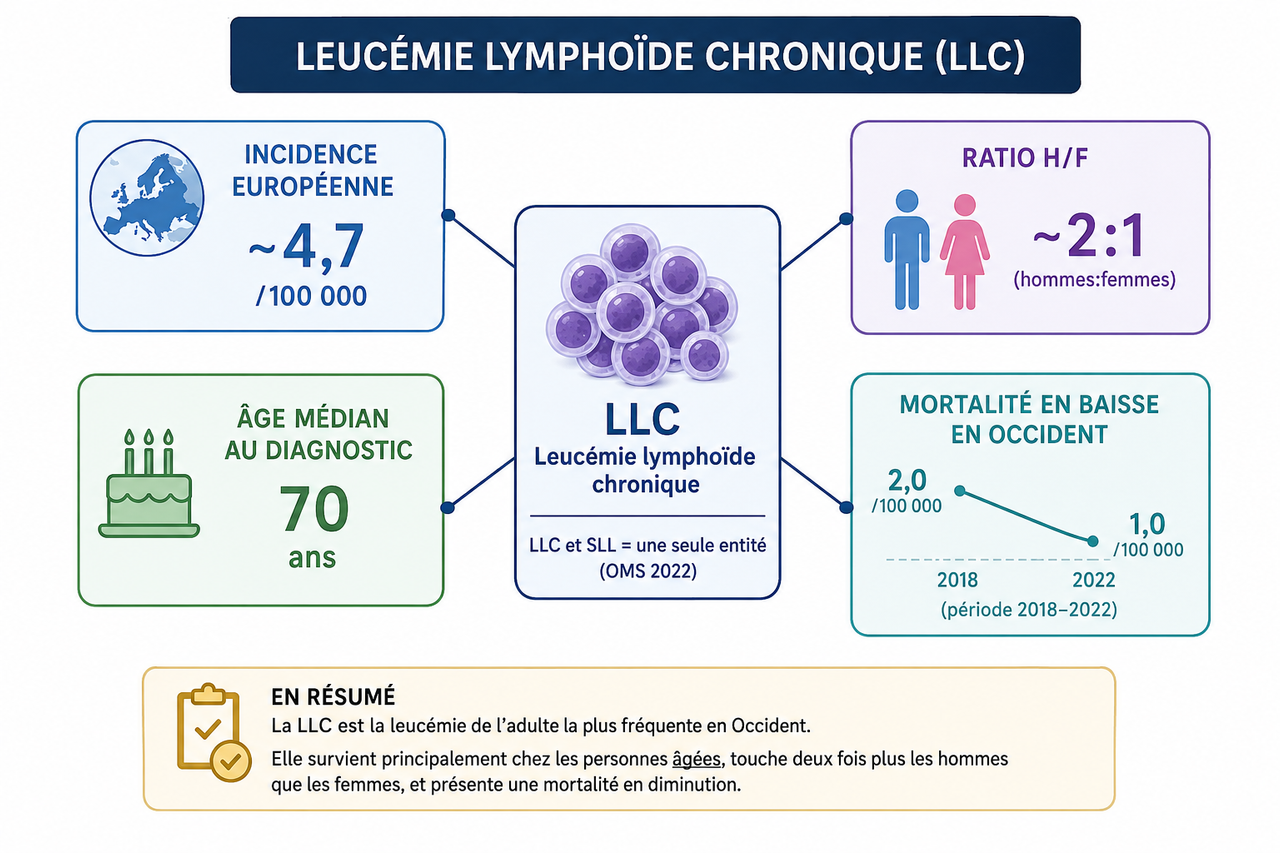
La LLC est la leucémie de l’adulte la plus fréquente en Occident, touchant surtout les sujets âgés, deux fois plus les hommes que les femmes, avec une mortalité en diminution.
3. Diagnostic, pathologie et biologie moléculaire
Le diagnostic repose sur l’immunophénotypage par cytométrie de flux, complété par les marqueurs additionnels lorsqu’un diagnostic différentiel se pose.
Phénotype LLC (cytométrie de flux)
Diagnostics différentiels
Statut IGHV
Le statut d’hypermutation somatique (SHM) des gènes IGHV dichotomise la maladie. La U-CLL (unmutated CLL, LLC à IGHV non muté ; ≥98 % d’identité germinale, ≤2 % de mutations) présente des caractéristiques génétiques souvent défavorables et une évolution rapide ; la M-CLL (mutated CLL, IGHV muté ; <98 % d’identité) correspond à une maladie indolente. Le statut IGHV prédit le temps jusqu’au 1ᵉʳ traitement et la durée de réponse selon les traitements, et reste stable dans le temps (contrairement aux aberrations FISH/cytogénétiques et à TP53). Les sous-sets stéréotypés #2 et #8 sont associés à une maladie agressive (données surtout rétrospectives). Séquençage Sanger ou NGS selon recommandations ERIC (couverture complète du VH CDR3, qui renseigne aussi l’appartenance au sous-set).
Aberrations de TP53
del(17p) (FISH) et/ou mutation TP53 (NGS préféré). Fréquence croissante : 5-10 % au diagnostic, 10-20 % avant 1ʳᵉ ligne, jusqu’à 50 % aux stades avancés et en transformation de Richter. Impact négatif indépendant sur la survie globale et la réponse à la chimio-immunothérapie ; aberrations souvent subclonales ou oligoclonales.
Sur le seuil de VAF : historiquement, un seuil arbitraire d’environ 10 % (hérité de la sensibilité du Sanger) servait à rapporter une mutation TP53 comme cliniquement significative. Le NGS détecte désormais des mutations à VAF <10 %, dont la pertinence clinique a été démontrée (étude multicentrique ERIC). Conséquence : le seuil de VAF pertinent sous thérapie ciblée reste indéfini, et s’appuyer sur les anciens seuils arbitraires de type Sanger n’est plus scientifiquement justifiable. À noter qu’une mutation à faible VAF peut représenter un clone majeur dans un prélèvement à faible fraction tumorale. Les agents ciblés, agissant indépendamment de p53, ne sont pas supposés accélérer l’expansion des clones TP53-déficients.
Autres aberrations
Les mutations SF3B1, NOTCH1, POT1, XPO1, NFKBIE restent hors routine (recherche). Le caryotype complexe (≥3 anomalies ; « high-CK » ≥5 = pronostic le plus défavorable) n’est pas recommandé hors essais (reproductibilité limitée).
Recommandations — bilan moléculaire
- Déterminer le statut IGHV (ERIC) chez tous les patients une seule fois avant la 1ʳᵉ ligne (stable). II, B
- Déterminer les aberrations de TP53 [del(17p) par FISH + mutation TP53 par NGS, ERIC] avant chaque ligne de traitement. II, B
- Évaluation du caryotype complexe non recommandée hors essais cliniques (reproductibilité limitée). III, B
Bilan diagnostique et pronostique selon le moment
4. Stadification et scores pronostiques
Deux systèmes cliniques, pronostiques au diagnostic, fondés uniquement sur l’examen clinique et la NFS (sans imagerie) : le Rai modifié (5 stades → 3 groupes de risque) et le Binet (3 groupes, plus utilisé en Europe).
| Binet | Rai | Risque | Définition |
|---|---|---|---|
| A | 0 | Faible | <3 aires lymphoïdes atteintes ; Hb ≥100 g/L et plaquettes ≥100 G/L |
| B | I–II | Intermédiaire | ≥3 aires lymphoïdes atteintes ; Hb ≥100 g/L et plaquettes ≥100 G/L |
| C | III–IV | Élevé | Anémie (Hb <100 g/L) et/ou thrombopénie (<100 G/L), quel que soit le nombre d’aires |
Les 5 aires lymphoïdes considérées : cervicale, axillaire, inguinale (uni- ou bilatérale comptant pour 1 chacune), rate, foie. La SLL est stadifiée selon la classification de Lugano, mais son évaluation du risque et son indication de traitement suivent celles de la LLC.
Suffixe A/B selon les symptômes B ; mention « bulky » pour une masse volumineuse.
⚠ Haut risque ≠ indication de traitement
Un classement haut risque (CLL-IPI, IPS-E, TP53, IGHV non muté) ne justifie pas de traiter une LLC asymptomatique précoce (Binet A/B). Aucun bénéfice de survie démontré, même avec agents ciblés (essai CLL12). On ne traite que sur critères iwCLL.
5. Indication de traitement (critères iwCLL 2018)
Le traitement n’est initié qu’en cas de maladie « active » selon iwCLL I, A (validé en treatment-naïf ; preuve plus limitée en rechute/réfractaire). Au moins un des critères suivants :
- Insuffisance médullaire progressive : apparition/aggravation d’une anémie et/ou thrombopénie. Seuils usuels Hb <100 g/L ou plaquettes <100 G/L (nuance : une thrombopénie stable au long cours n’impose pas à elle seule de traiter).
- Adénopathies massives (>10 cm) ou progressives/symptomatiques.
- Splénomégalie massive (>6 cm sous le rebord costal) ou progressive/symptomatique.
- Lymphocytose progressive : augmentation >50 % sur 2 mois, ou temps de doublement lymphocytaire <6 mois (après exclusion d’autres causes : infection, corticoïdes).
- Cytopénies auto-immunes (anémie/thrombopénie) mal contrôlées par les corticoïdes.
- Atteinte extranodale symptomatique ou compromettant un organe (peau, rein, poumon, rachis…).
- Symptômes liés à la maladie : perte de poids involontaire >10 % sur 6 mois ; fatigue significative (ECOG ≥2) ; fièvre >38,0 °C ≥2 semaines sans infection ; sueurs nocturnes >1 mois sans infection.
6. Surveillance active : prise en charge globale
Vaccinations
L’immunodéficience progressive est un marqueur de la LLC. Recommandés : pneumocoque (PCV20 au diagnostic, puis PPV23 ≥2 mois après ; rappel tous les 5 ans ; « catch-up » PCV20 si seulement PPV23 antérieur), VZV recombinant (Shingrix, sûr et immunogène même chez les patients lourdement prétraités ; (val)aciclovir en prophylaxie secondaire si antécédent de zona), VHB, grippe et SARS-CoV-2 (saisonniers, répétés selon recommandations nationales). Aucun vaccin vivant (ROR, polio oral, fièvre jaune, Zostavax). III, B
Anti-infectieux et immunoglobulines
Pas d’antibioprophylaxie systématique IV, D. Substitution en immunoglobulines si infections récurrentes/aiguës et hypogammaglobulinémie confirmée (IgG <4 g/L) ; voie sous-cutanée préférée (taux résiduels plus élevés, moins d’effets indésirables, meilleure qualité de vie) II, B.
Prévention et mode de vie
Risque accru d’autres cancers, même avant traitement : informer/encourager le dépistage population générale, le sevrage tabagique et la protection UV ; évaluation dermatologique chez les sujets à risque III, B. Vitamine D et compléments : pas de prescription routinière III, C.
Cytopénies auto-immunes (CAI)
5-10 % au cours de la maladie. Si la LLC ne remplit pas les critères iwCLL : traiter la CAI comme une CAI idiopathique ; si elle les remplit : traiter la LLC.
- AHAI à anticorps chauds : corticoïdes + rituximab supérieurs aux corticoïdes seuls ; si traitement LLC indiqué, iBTK ou inhibiteur BCL2 + anti-CD20.
- Maladie des agglutinines froides : réchauffement ; rituximab en monothérapie 1ʳᵉ ligne (corticoïdes peu efficaces).
- PTI (2-5 %) : corticoïdes ou IgIV (50-60 %), rituximab hebdomadaire ×4, eltrombopag ; traiter la LLC avant une 2ᵉ/3ᵉ ligne de PTI.
- Érythroblastopénie / neutropénie auto-immune (<1 %) : même approche que l’AHAI.
Recommandation : LLC à traiter → traitement LLC standard ; LLC sans indication → corticoïdes ± Ig, puis rituximab ou démarrage du traitement LLC en cas de réponse insuffisante IV, A.
7. Traitement de première ligne
Facteurs guidant la sélection
- Profil biologique : aberrations TP53 et statut IGHV
- Préférence du patient et décision partagée
- ECOG et comorbidités, surtout cardiovasculaires (HTA)
- Durée et mode d’administration (oral vs perfusion, durée limitée vs continu)
- Profil de toxicité de chaque agent
- Interactions médicamenteuses, notamment vénétoclax (CYP3A : azolés, macrolides) — adapter aussi en cas d’insuffisance rénale/hépatique ou polymédication
- Inclusion dans un essai clinique à proposer dès que possible
Résultats des principales études de 1ʳᵉ ligne
| Schéma | Étude (phase) | Comparateur | Population | Résultat principal |
|---|---|---|---|---|
| Vénétoclax + obinutuzumab | CLL14 (III) | Chlorambucil + obinutuzumab | Naïfs, comorbidités | PFS médiane 76,2 mois ; PFS 6 ans 53 % ; TTNT 6 ans 65 % ; HR 0,40 |
| Vénétoclax + obinutuzumab | CLL13/GAIA (III) | FCR ou BR (CIT) | Naïfs fit | PFS 5 ans 81,8 % (vs CIT 62,0 %) |
| Vénétoclax + obi + ibrutinib (triplet) | CLL13/GAIA (III) | FCR ou BR (CIT) | Naïfs fit | PFS 5 ans 85,5 % ; infections G3-4 21 % |
| Vénétoclax + ibrutinib | GLOW (III) | Chlorambucil + obinutuzumab | ≥65 ans / unfit | PFS 59,9 % à 64 mois |
| Vénétoclax + ibrutinib | CAPTIVATE (II) | — (bras unique) | ≤70 ans | PFS 5,5 ans 66 % ; OS 5,5 ans 97 % |
| Vénétoclax + ibrutinib (guidé MRD) | FLAIR (III) | Ibrutinib seul ; FCR | Naïfs | PFS 5 ans 93,9 % (vs ibrutinib 79,0 % ; FCR 58,1 %) |
| Acalabrutinib + vénétoclax ± obinutuzumab | AMPLIFY (III) | FCR ou BR (CIT) | Naïfs, TP53 sauvage | PFS 36 mois 76,5 % (doublet) / 83,1 % (triplet) vs CIT 66,5 % ; OS 36 mois 94,1 % (doublet) |
| Ibrutinib | RESONATE-2 (III) | Chlorambucil | ≥65 ans | PFS médiane 8,9 ans (suivi 10 ans) |
| Acalabrutinib ± obinutuzumab | ELEVATE-TN (III) | Chlorambucil + obinutuzumab | Naïfs | PFS 72 mois 78 % (Acala-Obi) / 62 % (Acala) vs CIT 17 % |
| Zanubrutinib | SEQUOIA (III) | Bendamustine + rituximab (BR) | Naïfs | PFS 5 ans 76 % |
| Ibrutinib | PCYC-1102 (Ib/II) | — (bras unique) | TP53 altéré, naïf | PFS médiane 53 mois |
| Vénétoclax + ibrutinib | CAPTIVATE (II) | — (bras unique) | del(17p)/TP53 | PFS 5,5 ans 30 % (47 % si MRD indétectable) |
| Zanubrutinib + vénétoclax | SEQUOIA arm D (II) | — (bras unique) | TP53 aberrant | PFS 3 ans 88 % (combinaison non autorisée) |
Sources : CLL14 — Al-Sawaf et al., Blood 2024;144:1924-1935. CLL13/GAIA — Fürstenau et al., Lancet Oncol 2024;25:744-759. GLOW — Niemann et al., Lancet Oncol 2023;24:1423-1433. CAPTIVATE — Wierda/Allan et al., HemaSphere 2025;9:e70151. FLAIR — Munir et al., N Engl J Med 2025;393:1177-1190. AMPLIFY — Brown et al., N Engl J Med 2025;392:748-762. RESONATE-2 — Burger et al., Blood 2025;146:2168-2176. ELEVATE-TN — Sharman et al., Blood 2025;146:1276-1285. SEQUOIA — Shadman et al., JCO 2025;43:780-787 ; arm D — Shadman et al., JCO 2025;43:2409-2417. PCYC-1102 — Byrd et al., Clin Cancer Res 2020;26:3918-3927.
Grille intégrée de choix (d’après la guideline, Figure 2)
| Schéma | IGHV muté | IGHV non muté | TP53 / del(17p) | Risque infect. | Facilité | Cardio- pathie | Insuff. rénale | Anticoag/ saign. |
|---|---|---|---|---|---|---|---|---|
| Vénétoclax + obinutuzumab | A | A | B | C | C | A | B | B |
| Vénétoclax + ibrutinib¹ | A | A | B | B | B | C | B | B |
| Vénétoclax + acalabrutinib | A | A | B | B | B | B | B | B |
| Véné + acala + obinutuzumab | A | A | B | C | C | C | B | B |
| Acalabrutinib | B | A | A | A | A | B | A | B |
| Zanubrutinib | B | A | A | A | A | B | A | B |
| Ibrutinib | B | A | A | A | A | C | A | B |
Interactions médicamenteuses : à évaluer au cas par cas pour tous les schémas. ¹ Particulièrement efficace en IGHV non muté dans une stratégie guidée par MRD, mais non approuvé. Prudence si ClCr <50 mL/min (vénétoclax) et <30 (iBTK), prévention du SLT si ClCr 50-79 ; iBTK contre-indiqués avec les anti-vitamine K.
Recommandations — 1ʳᵉ ligne
- La chimio-immunothérapie n’est plus recommandée (inférieure ou risque de néoplasies myéloïdes secondaires avec FCR). I, A
- Traitement à durée limitée préféré au continu hors aberration TP53. I, A
- En IGHV muté, éviter le traitement continu. II, A
- Acalabrutinib/zanubrutinib préférés à l’ibrutinib pour le continu (ne pas switcher un patient stable et bien toléré sous ibrutinib). I, A
- En cas d’aberration TP53, traitement continu par iBTK suggéré (sauf préférence partagée pour le durée-limitée). I, A
8. Traitement de la rechute / réfractaire
Ne traiter qu’en cas de critères iwCLL remplis. Le choix dépend du traitement antérieur (durée limitée vs continu), de la durée de réponse, du motif d’arrêt (progression vs intolérance), du statut TP53 et IGHV. L’inclusion en essai clinique est à proposer activement.
Après chimio-immunothérapie : pas de re-chimio si options ciblées accessibles → vénétoclax + rituximab ou iBTK 2ᵉ génération I, A. Après iBTK covalent : si progression, basculer vers vénétoclax ou pirtobrutinib (ne jamais ré-exposer à un autre iBTK covalent) II, B. Après durée limitée par vénétoclax : re-traitement vénétoclax si réponse durable et intervalle >2 ans, sinon changer de classe IV, B. Double-réfractaire : pirtobrutinib préféré (ou idélalisib + rituximab), comme pont vers l’alloCSH chez les éligibles II/III, A ; éviter d’interrompre le traitement en cours avant la ligne suivante III, B.
9. Transformation de Richter (RT)
Lymphome agressif survenant chez 2-10 % des patients LLC. Biopsie excisionnelle idéalement, pour le diagnostic et l’exclusion des différentiels (LLC accélérée, pseudo-RT sous arrêt d’iBTK, progression prolymphocytaire). La TEP-FDG guide la biopsie. L’enjeu central est la relation clonale (séquençage IGHV) : la RT clonalement reliée (~80 %) a un pronostic défavorable.
La RT-DLBCL non reliée se traite comme un DLBCL de novo ; la RT-cHL comme un Hodgkin de novo. Pour la RT-DLBCL reliée ou de relation non établie : pas de standard, essais cliniques préférés ; hors essais, chimio-immunothérapie type R-CHOP (efficacité limitée : RC 5-38 %, OS médiane <1 an). Pistes en phase 2 : pirtobrutinib (ORR ~50 %), vénétoclax + R-DA-EPOCH (RC 50 %), combinaisons checkpoint (RT1 : zanubrutinib + tislélizumab 58 % ; MOLTO : obinutuzumab-atézolizumab-vénétoclax 68 %), bispécifiques (epcoritamab 48 %), CAR-T anti-CD19 (RC ~46 %). AlloCSH en consolidation chez les patients fit avec RT-DLBCL reliée en rémission III, B.
Fiche synthétique — LLC & Richter (EHA 2026)
Diagnostic & bilan
- CMF : ≥5×10⁹/L B clonaux CD19+/CD5+/CD23+, CD20/CD79B/Ig faibles, restriction κ/λ.
- IGHV (ERIC) : une fois avant 1ʳᵉ ligne (stable). TP53 [del(17p) FISH + mutation NGS] : avant chaque ligne. Caryotype complexe hors routine.
- Seuil de VAF TP53 sous thérapie ciblée indéfini ; anciens seuils Sanger non justifiables.
Quand traiter
- Uniquement sur critères iwCLL 2018 (cytopénies, adénopathies/splénomégalie volumineuses ou évolutives, TDL <6 mois, CAI réfractaire, symptômes B).
- Le haut risque biologique seul n’est pas une indication de traiter.
1ʳᵉ ligne
- Chimio-immunothérapie abandonnée.
- Sans TP53 : durée limitée préférée. IGHV muté → Véné+Obi (éviter le continu). IGHV non muté → Acala+Véné±Obi ou Véné+Obi.
- TP53/del(17p) : continu par iBTK 2ᵉ gén. (acala/zanu > ibrutinib) ; triplets si tolérance.
- iBTK 2ᵉ génération préférés ; ne pas switcher un patient stable sous ibrutinib. BCL2 favorisé si cardiopathie/anticoagulation.
Rechute / réfractaire
- Après CIT : Véné-R ou iBTK 2ᵉ gén.
- Après iBTK covalent (progression) : vénétoclax ou pirtobrutinib — jamais un autre iBTK covalent.
- Après vénétoclax : re-traiter si intervalle >2 ans, sinon changer de classe.
- Double-réfractaire : pirtobrutinib (ou idélalisib+R), pont vers alloCSH ; essais cliniques.
Richter
- Biopsie excisionnelle + relation clonale (IGHV) + TP53.
- Non relié → DLBCL de novo ; cHL → Hodgkin de novo ; relié → essai clinique > R-(DA)-EPOCH, alloCSH si réponse.
Source principale : Eichhorst B, Ghia P, et al. EHA Guidelines on management of chronic lymphocytic leukemia and Richter transformation. HemaSphere 2026;10:e70403. Synthèse à visée pédagogique — ne se substitue pas au texte intégral des recommandations.